In this summer, I have worked on a project to develop tools that make BioJulia run faster. As an outcome, Automa.jl now generates more efficient code to parse text files, ConcurrentCalls.jl runs multiple tasks in parallel, and simple and efficient interfaces to various compression formats are provided in TranscodingStreams.jl.
Instruction-level parallelism
Instruction-level parallelism is a technique to run multiple instructions simultaneously on a CPU core. This may sound odd but actually it is possible because CPU instructions are executed in multiple stages such as fetch, decode, execute, and so on. This is called instruction pipelining and modern processors can execute these stages in parallel and hence processing throughput may be boosted by utilizing it.
I implemented automatic loop unrolling in Automa.jl, which is a package to generate finite state machine (FSM) from regular expressions. We use it to generate high-performance parsers for various file formats used in bioinformatics. The loop unrolling factor can be specified as an argument of Automa.CodeGenContext. For example, you can specify the value 8 in this way: context =
Automa.CodeGenContext(loopunroll=8) and that is all. The code generator of Automa.jl will automatically unroll loops found in an FSM.
Automa.jl works by incrementally increasing a reading position variable and reading data byte by byte from a buffer. This is difficult to unroll because the position variable creates a critical path of data dependencies. So, Automa.jl unrolls particular nodes in an FSM that have a self edge and have no actions within state transition.
Let's see the improvement of performance. FASTA and FASTQ are one of the most common file formats to store biological sequences. I benchmarked parsing throughput in these two formats. The throughput improved as the loop unrolling factor was increased and saturated around factor = 10.
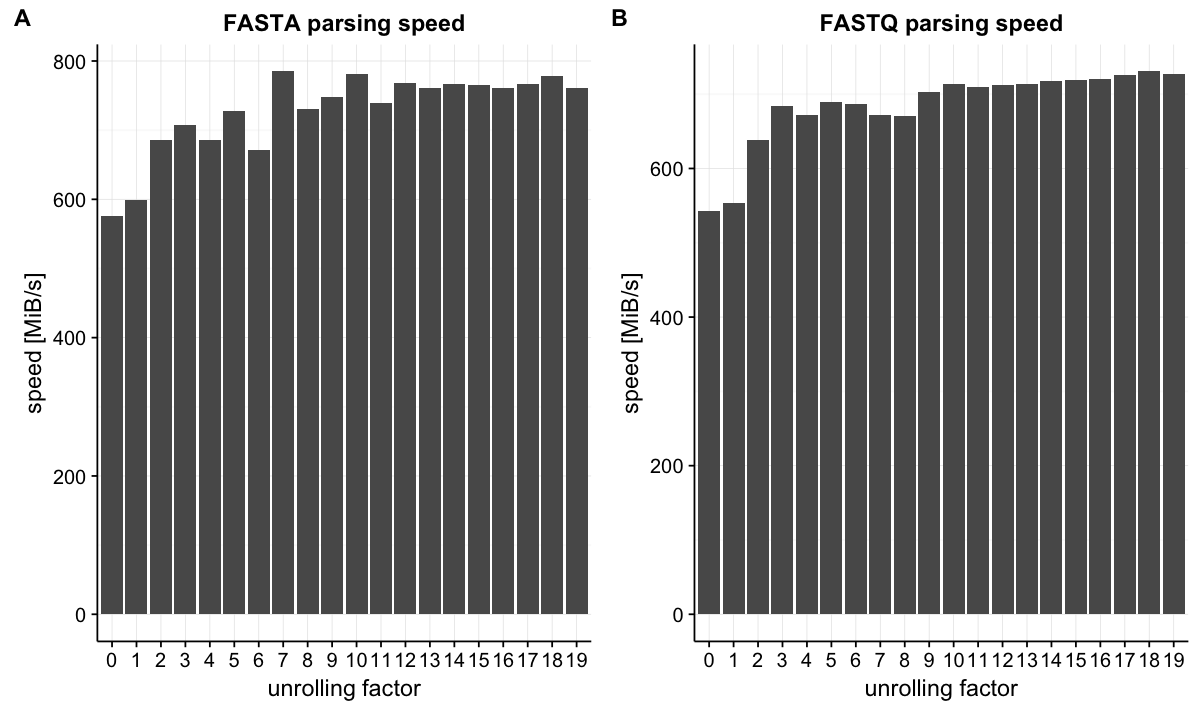
Unrolled parsing achieved about 1.3 times speedup in both cases. This benchmark does not include I/O operations but other operations that are required to convert byte data to our data type are included. The improvement may not look surprising, however, we should note that it is achieved almost for free. There is no need to use more CPU cores.
Task-level parallelism
ConcurrentCalls.jl is a package that makes function calls (or tasks) run concurrently. It exports a macro @cc that transforms Julia code to code that calls functions on remote processes. This is a conceptual example of running tasks in parallel:
addprocs(2)
using ConcurrentCalls
function multitask()
# Define some time-consuming (say >10ms) task(s).
function sum(x, y)
sleep(2)
return x + y
end
function prod(x, y)
sleep(1)
return x * y
end
# Call functions concurrently using multiple processes.
@cc begin
x = sum(1, 2) # => 3
y = sum(3, 4) # => 7
prod(x, sum(y, 1)) # => 24
end
end
multitask() # => 24In the example above, x = sum(1, 2) and y = sum(3, 4) will be executed in parallel on different processes. These function calls are arranged by a scheduler of ConcurrentCalls.jl and each taks is assigned to a worker process based on some heuristics. The third line inside the @cc macro has two function calls. Each of them will also be assigned to a worker but it never starts until the first two finish. Dependencies of tasks are handled by the scheduler and a task will not be executed until all of its arguments are finished and results are available.
Execution of tasks are defined in an imperative way and so its semantics would be apparent from the code. Data dependencies are naturally expressed by arguments passed to functions. The code below shows three examples that use a for loop to describe a job:
# independent tasks
@cc begin
for i in 1:100
task()
end
end
# sequential taks (not parallelizable)
@cc begin
a = task1()
for i in 1:100
a = task2(a)
end
end
# biparallel tasks
@cc begin
a = task1()
b = task2()
for i in 1:100
a = task3(a)
b = task4(b)
end
endThis package is still at a very early stage of development but some simple subset of Julia code can be executed in parallel. See the README page of ConcurrentCalls.jl for more examples and some caveats.
New I/O APIs
A difficulty of parallelizing data processing in bioinformatics is that input files are often compressed. The most common compression format, gzip, does not support either parallel decompression or splitting data into smaller chunks. Some special file formats (BGZF for example) can support parallel decompression and chunking but gzip is still slow.
A recent compression algorithm known as Zstandard (or Zstd) is much faster than gzip while keeping the compression ratio at the same level of gzip. Moreover, it started to implement an experimental support of seekable compression format, in which data are split into frames and each of which are compressed independently. Tables to seek to a specific position are stored in skippable frames and hence it can support parallel processing and chunking.
To support this feature, I implemented an interface package to Zstd, which is already available as CodecZstd.jl. While it does not yet support the seekable format, we can enjoy the performance of Zstd immediately. Also, this package is built on top of TranscodingStreams.jl, which offers simple and consistent APIs for many data formats. Currently, gzip (zlib), bzip2, xz, and zstd are supported. These packages are replacements of Libz.jl and BufferedStreams.jl I maintain.
Plans
Speeding up programs never ends. I still have some pending pull requests that are required to work well with these tools (https://github.com/BioJulia/BioCore.jl/pull/7, https://github.com/BioJulia/BioAlignments.jl/pull/4, https://github.com/BioJulia/BioSequences.jl/pull/25). ConcurrentCalls.jl lacks examples, tests, benchmarks, and many features to be used in real work. CodecZstd.jl will support the seekable format once it gets stable. I really welcome feedbacks of these designs and ideas to improve them further.
Acknowledgments
I appreciate contributions of my mentor Shashi Gowda and other members of the Julia community.